It was initially thought that type I IFN
and type III IFN performed largely redundant functions. However, it is
becoming increasingly clear that type III IFN exert distinct and
non-redundant functions compared to type I IFN, especially in mucosal
tissues. Here, we review recent progress made in unraveling the role of
type I/III IFN in intestinal mucosal tissue in the steady state, in
response to mucosal pathogens and during inflammation.
Introduction
The intestinal tract is a major entry site for viruses
and bacteria. Mucosal innate and adaptive immune cells are equipped to
respond to and fight invading pathogens. At the same time, the
intestinal lumen is densely colonized by commensal microflora, which at
steady state does not provoke an exacerbated inflammatory response.
The
intestinal lumen is separated from the underlying sterile
lamina propria
harboring the body’s largest immune cell compartment by a single layer
of
polarized intestinal epithelial cells (IECs). This epithelial cell
layer undergoes rapid and perpetual self-renewal without disrupting the
functional integrity of cell–cell junctions. In addition, IECs not only
form a passive physical barrier but also participate actively in the
immune response against major enteric pathogens and cross talk with the
commensal flora (
1,
2). However, pathogenic viruses, bacteria, and parasites exploit opportunities for breaching the epithelial barrier.
Upon infection, host cells communicate by means of
production and secretion of signaling molecules. Interferons (IFN) are a
large family of cytokines with diverse functions during a successful
host defense.
The family of type I IFN comprises more than 20 members
with multiple IFN-α and one IFN-β being the most important. Classically,
the most prominent function of type I IFN is to induce antiviral
immunity, whereas IFN-γ, the only type II IFN, promotes the response to
intracellular bacteria. However, a vast amount of studies has found that
type I IFN are also produced during bacterial infection. In contrast to
their action in viral infections, their activity against bacteria can
be either favorable or detrimental for the host (
3–
6).
Recently, a novel family of IFN, the type III IFN or IFN-λ family, was described (
7,
8).
This family consists of IFN-λ1, IFN-λ2, IFN-λ3 (also called IL-29,
IL-28A, and IL-28B), and IFN-λ4 in humans, whereas mice have only two
functional genes encoding IFN-λ (
Ifnl2 and
Ifnl3) and two
Ifnl1 pseudogenes (
9).
Similar to type I IFN, type III IFN are induced by viral infection and
show antiviral activity. However, they are
structurally distinct from
type I IFN and interact with a heterodimeric class II cytokine receptor
consisting of the
IFN-λR1 (also called
IL-28Rα) chain in complex with
the
IL-10R2 chain, opposed to the type I IFN receptor (IFNAR).
A number of studies have addressed the functional
importance of type III IFN compared to type I IFN in the context of
viral infections (
10–
15).
Less is known about the role of type I IFN and almost nothing on the
role of type III IFN in the host defense against bacterial
enteropathogens, intestinal homeostasis, and colitis. Therefore, we
review recent progress made on the importance of type I and III IFN
during
enteric viral infections and focus on the role of type I IFN in
the intestinal mucosal tissue during steady state, in response to
bacterial infections and during inflammation.
Induction of Type I and III IFN
The induction of type I and III IFN has been recently reviewed elsewhere (
16),
therefore we will only briefly summarize the
major mechanism leading to
IFN expression. Virtually all cells are equipped with the machinery to
recognize viral infection and express type I and III IFN in response.
Similar stimuli and pathways lead to the expression of type I and III
IFN; however, differences between cell types as well as in magnitude and
kinetics have been described (
14,
16,
17).
Comparable expression patterns of type I and III IFN result from a
similar requirement of transcription factors for the expression of their
encoding genes, such as
IFN regulatory factors (IRFs) and
NF-κB. There
are however
some differences in the promoter region, with
IFN-β
expression relying on the binding of the constitutively expressed
IRF-3
to its promoter, which allows rapid induction. By contrast,
IFN-α
requires IRF-7 binding, which is an interferon-stimulated gene (ISG)
itself and needs to be upregulated in most cell types following
infection (
3). Type III IFN are more dependent on the activation of NF-κB (
18) and require the combined action of IRFs and NF-κB for full induction (
19–
21).
During
systemic viral infections hematopoietic cells are
the major source of type I IFN. Plasmacytoid dendritic cells (pDCs),
which are designated as being the “professional” type I IFN-producing
cells, produce large amounts in response to a wide range of viruses,
parasites, and bacteria and are particularly important in the
early
phase of type I IFN production (
22–
24).
However, depending on the infectious agent,
myeloid cells are also
involved in systemic type I IFN production. During systemic
Listeria
infection, the vast amount of systemic IFN-β production is independent
of pDCs but seems to be produced by LysM-Cre-expressing
macrophage/monocyte-like cells including TipDCs but not neutrophils (
25–
27).
In the intestinal lamina propria, dendritic cells (DCs) as well as
mononuclear phagocytes produce IFN-β and IFN-α5 in the steady state (
14,
23,
28).
Epithelial cells are thought to be the major producer of
type III IFN at steady state and during enteric viral infection, while
lamina propria leukocytes (LPLs) also produce type III IFN under certain
conditions (
14,
29).
Intraepithelial lymphocytes (IEL) produce IFN-α and IFN-λ upon TCR
activation, which contributes to protection during
norovirus infection (
30).
Moreover, Th17 cells are the main source of IFN-λ in
psoriatic lesions of the skin (
31).
Bacteria trigger similar intracellular signaling cascades
to viral infections and many bacterial infections lead to the
production of type I IFN [reviewed in Ref. (
32,
33)].
Induction of type III IFN has been demonstrated only for a limited
number of bacterial species. A human epithelial
colon cancer cell line
expresses type III IFN upon infection with Gram-positive bacteria such
as
Listeria monocytogenes (
34,
35),
Staphylococcus aureus, and Enterococcus faecalis but fails to produce considerable amounts of type III IFN when infected with Gram-negative bacteria such as
Salmonella enterica ssp. Typhimurium,
Shigella flexneri, and
Chlamydia trachomatis (
35). Induction seems to be cell type, species, and gene specific (
36,
37).
Signaling in Response to IFN
Binding of IFN to their corresponding receptors triggers
the stimulation of a
Janus kinase (JAK)–
signal transducer of
transcription (STAT) pathway. The type I IFN receptor (IFNAR) consists
of two subunits, IFNAR1 and IFNAR2. Engagement of IFNAR with its ligand
ultimately results in the activation of the transcription factor complex
ISGF3 comprised of STAT1/STAT2 heterodimers in conjunction with IRF-9
and subsequently the induction of ISGs (
3,
38).
The type III IFN receptor consists of the unique IFN-λR1
chain and the IL-10R2 chain, which is shared with the IL-10 receptor.
Engagement of this
receptor complex results in the activation of a
signal transduction cascade in a manner highly similar to that caused by
type I IFN signaling. Interestingly, signaling by type III IFN is
additionally regulated at the level of receptor expression. Whereas
IFNAR is ubiquitously present, the
IFN-λR1 chain of the type III IFN
receptor is only expressed in a limited number of cell types,
preferentially located at
mucosal surfaces. Epithelial cells in mucosal
tissues are a major target of type III IFN (
39,
40). Additional responsiveness to type III IFN has recently been suggested for a restricted panel of immune cells (
9). Type III IFN was proposed to have a role in the
direct regulation of NK cell effector function (
41).
A suppressive function of type III IFN in autoimmune and inflammatory
diseases was also proposed recently. In a model of collagen-induced
arthritis, treatment with type III IFN inhibits the recruitment of
IL-1β-expressing neutrophils, which have been shown to express high
levels of IFN-λR1 and respond directly to type III IFN (
42). In addition, there are controversial data on the responsiveness of T cells, DCs, and monocytes to type III IFN (
9).
In human cells, expression of the type III IFN receptor seems to be
less restricted than in mouse cells and a wider panel of immune cells,
including B cells, is responsive to type III IFN (
43).
Signaling by IFNs induces the transcription of hundreds
of ISGs. These include pattern-recognition receptors, antiviral
effectors such as myxovirus resistance (Mx) gene 1 and 2, pro-apoptotic
genes, MHC class I genes, inducible nitric oxide synthase, and genes
encoding members of the GTPase superfamily which alter the maturation of
phagosomes to counteract pathogen strategies based on survival in
intracellular compartments.
Moreover genes involved in the
desensitization to IFNs are also induced,
allowing cells to recover
from
the IFN response (
38,
44).
The importance of IFNs in the immediate defense against pathogens has
been shown by the generation of gene-targeted mice. Mice deficient in
components of the type I IFN signal transduction pathway are highly
susceptible to a variety of viruses (
5,
45).
The role of type I IFN in bacterial infections is more complex. Whereas
type I IFN protect mice against systemic infection with most
extracellular bacteria tested, they
exacerbate disease during infection
of mice with
L. monocytogenes or
Mycobacterium tuberculosis (
3–
6).
Enteric Viral Infections and IFN
Studies investigating the functional importance of type I
IFN versus type III IFN in the context of systemic viral infections
found
a dominant phenotype for type I IFN and
only a small contribution
of type III IFN in the absence of type I IFN. The first indication for a
tissue-specific role of type III IFN arose from studies with
organ-tropic viral infections suggesting that type III IFN are important
in enforcing and strengthening the antiviral response at mucosal sites
(Table
1) (
10–
12,
46–
48). The gastrointestinal tract, lung, vagina, and salivary glands respond strongly to systemic IFN-λ expression (
40).
In the lung and
gastrointestinal tract, epithelial cells were
identified to express high levels of the
type III IFN receptor and
represent the major target of type III IFN (
11).
These findings explain why mice deficient for both IFN systems are more
susceptible to lung-tropic viruses, such as influenza A and B virus,
respiratory syncytial virus, and severe acute respiratory syndrome
coronavirus than single type I IFN receptor-deficient mice (
11). The remaining part of this section focuses on the role of type III IFN in enteric viral infections.
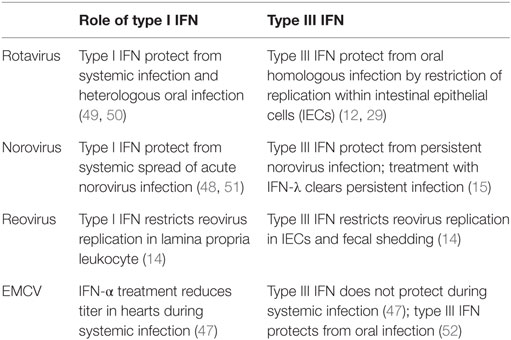
Rotavirus
Rotavirus belongs to the family of reoviridae and
infection of humans leads to severe diarrhea in children younger than 5
years. The susceptibility of infants can be recapitulated in a mouse
model, where suckling mice are highly susceptible to infection compared
to adult mice. The strict host cell tropism of rotavirus for IECs makes
it a clean model to study epithelial-specific effects of IFN.
Mice can be infected with a homologous strain of murine
rotavirus or with a heterologous strain such as rhesus or simian
rotavirus. Homologous strains are better equipped to evade the host
immune response, which generally leads to higher viral titers and a more
severe pathology at a lower infectious dose (
49).
A protective role of type I IFN and IFN-γ has been
questioned, since mice impaired in type I IFN or IFN-γ signaling
infected with a murine rotavirus strain do not show differences in viral
load, and treatment with either type I IFN or IFN-γ did not result in a
clinical benefit (
53).
However, simian and rhesus rotavirus show enhanced systemic replication
in mice deficient for type I IFN and IFN-γ receptor or STAT1.
By contrast, type III IFN were protective in a homologous infection model of suckling and adult mice (
12).
Of note, a very distinct cell tropism for type III IFN responsiveness
in the intestine was reported: IECs were solely activated by type III
IFN and are not responsive to type I IFN, whereas cells in the lamina
propria respond to type I IFN induced during viral infection (
12).
Supporting these findings it was shown that IL-22 augments the
antiviral effects of type III IFN signaling and contributes to the
protective effect during homologous rotavirus infection (
29).
However, this model has been questioned by another study reporting type
I IFN- and type III IFN-mediated protection only for heterologous but
not for homologous rotavirus infection of suckling mice (
50).
Experimental discrepancies between those studies are not apparent
suggesting that flora differences between mouse facilities or genetic
strategy of the knock-out mouse lines might impact on the efficacy of
IFN signaling. Of note, Lin et al. reported age-dependent responsiveness
of IECs toward IFNs with neonatal IECs being responsive to both type I
IFN and type III IFN, whereas adult IECs were responsive to type III IFN
only (
50).
Norovirus
Norovirus is the cause of the majority of non-bacterial
gastroenteritis in adults. In contrast to rotavirus, the host cell
tropism of norovirus is broad and not fully characterized.
Ex vivo and most
in vivo studies could not show productive virus replication in IECs (
54). Phagocytes allow productive virus replication and during
in vivo infection, virus was detected in LPLs (
54,
55).
Although the virus does not replicate in IECs, it has been suggested
that it translocates across the epithelium or enters the host
via M cells (
56).
Type I IFN and IFN-γ restrict murine norovirus replication in macrophages and DCs
in vitro (
57,
58).
In vivo, the antiviral activity of type I IFN mediates some protection from systemic replication of an acute strain (
51) and after high-dose oral infection (
59).
However, local replication in the colon and fecal shedding of a
persistent norovirus strain is controlled by type III IFN. Treatment
with type III IFN resolves persistent infection, independent of adaptive
immune responses, by acting on non-hematopoietic cells (
15).
By contrast, type I IFN controls the systemic spread and persistency of
the acute norovirus strain CW3 by activation of the host DCs (
48).
These findings demonstrate the distinct cell-type specificities of type
I IFN and type III IFN during infection: local protection in the colon
through type III IFN stimulation of epithelial cells and prevention of
systemic spread and persistency by type I IFN in myeloid cells.
The commensal bacterial flora was reported to promote
norovirus persistency in the intestine and antibiotic treatment of mice
prevented persistent infection with norovirus. The protective effect was
only observed in the presence of functional type III IFN signaling (
13).
The antibiotic treatment did not alter type III IFN signaling and
therefore the authors concluded that the microflora might render the
virus susceptible to the antiviral action of type III IFN.
Alternatively, the absence of type III IFN signaling might increase the
host’s vulnerability to persistent viral infection so dramatically that
minor changes by antibiotic treatment do not impact on the overall
susceptibility under those conditions.
Reovirus
Reovirus has a broad host cell tropism and replicates in
epithelial cells and immune cells of the intestinal mucosa. After oral
infection, it enters the host
via M cells into Peyer’s patches
and can spread further during infection. Type I IFN produced by
hematopoietic cells is essential to limit systemic spread of the virus
and to prevent lethality (
60).
In a study using type I IFN or type III IFN signaling-deficient mice,
it was demonstrated that type III IFN signaling specifically prevents
replication of the virus in IECs, whereas type I IFN signaling limits
replication in lamina propria cells and systemic spread of the virus (
14).
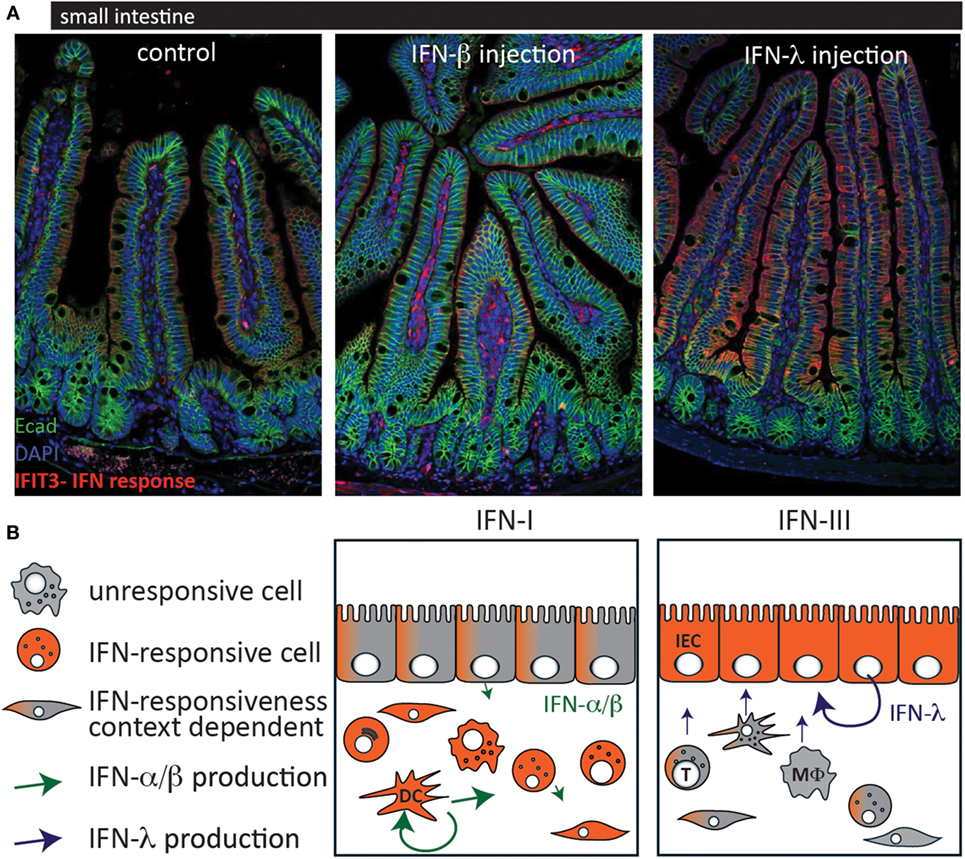
This study confirms the compartmentalized action of IFN in the
intestinal mucosa and provides an explanation by showing that IECs only
express low levels of IFNAR (Figure
1).
Furthermore, it was shown that the production of IFN is cell type
specific in that IECs produce higher levels of type III IFN and LPLs
predominantly produce type I IFN.
Taken together, the studies of
enteric viral models with rotavirus, reovirus, and norovirus show a
strong and specific responsiveness of IECs to type III IFN (
12,
15,
29).
Therefore, type III IFN might specifically enforce the intestinal
barrier against enteric viruses and also against viral entry via
the intestinal route. Additionally, a strong IFN response by type III
IFN signaling within the epithelial lining prevents viral spreading (
12,
14,
15). Studies showing that type III IFN treatment protects against oral EMCV (
52) infection but not from systemic infection (
47) support the conclusion that type III IFN protects the host not only from enteric viruses but also from viral entry
via
the oral route. By contrast, the contribution of type I IFN to the
epithelial antiviral response in the intestine is less clear and
conflicting results suggest it to be context dependent (
12,
29,
50).
----- ( Artikkelissa mainitaan usea bakteeritulehdus)
Inga kommentarer:
Skicka en kommentar