Viruses. 2019 Feb 20;11(2). pii: E171. doi: 10.3390/v11020171.
Influenza Virus Infections and Cellular Kinases.
Abstract
Influenza
A viruses (IAVs) are a major cause of respiratory illness and are
responsible for yearly epidemics associated with more than 500,000
annual deaths globally. Novel IAVs may cause pandemic outbreaks and
zoonotic infections with, for example, highly pathogenic avian influenza virus (HPAIV) of the H5N1 and H7N9
subtypes, which pose a threat to public health. Treatment options are
limited and emergence of strains resistant to antiviral drugs jeopardize
this even further. Like all viruses, IAVs depend on host factors for
every step of the virus replication cycle. Host kinases link multiple
signaling pathways in respond to a myriad of stimuli, including viral
infections. Their regulation of multiple response networks has justified
actively targeting cellular kinases for anti-cancer therapies and
immune modulators for decades. There is a growing volume of research
highlighting the significant role of cellular kinases in regulating IAV
infections. Their functional role is illustrated by the required
phosphorylation of several IAV proteins necessary for replication and/or
evasion/suppression of the innate immune response. Identified in the
majority of host factor screens, functional studies further support the
important role of kinases and their potential as host restriction
factors. PKC, ERK, PI3K and FAK, to name a few, are kinases that
regulate viral entry and replication. Additionally, kinases such as IKK,
JNK and p38 MAPK are essential in mediating viral sensor signaling
cascades that regulate expression of antiviral chemokines and cytokines.
The feasibility of targeting kinases is steadily moving from bench to
clinic and already-approved cancer drugs could potentially be repurposed
for treatments of severe IAV infections. In this review, we will focus
on the contribution of cellular kinases to IAV infections and their
value as potential therapeutic targets.
KEYWORDS:
antivirals; influenza virus; kinases; metabolism; pathogenesis; phosphorylation; replication; small molecule inhibitors- PMID:
- 30791550
- DOI:
- 10.3390/v11020171
Free full text

The successful nuclear export of vRNP has been shown to depend directly on the viral activation of the Raf/MEK/ERK signaling pathway [31,109].
MAPK kinase (MEK) and extracellular signal-regulated kinase (ERK), belong to the group of classical mitogen-activated protein kinases (MAPK). MEKs have been shown to regulate IAV and IBV replication [31,109]. Several MEK inhibitors resulted in vRNP retention, reduced titers of progeny virus in vitro, and also improved mouse survival in vivo [57,58,59].
During early stages of IAV infection, ERK regulates the vacuolar H+-ATPase (V-ATPase) activity to mediate pH-dependent acidification of endosomes and subsequent fusion of the viral and endosomal membranes [41]. In vitro inhibition of ERK, a direct downstream mediator of MEK, impedes IAV vRNP nuclear import as well as export [41,60].
IAVs activation of Raf/MEK/ERK signaling also induces p90 ribosomal s6 kinases (RSK), which play an important role as downstream mediators of ERK signaling [61,110]. RSK2 is involved in regulation of cell growth and proliferation. RSK2 knockdown using shRNAs results in increased IAV and IBV replication and IAV polymerase activity [61]. Inhibition of RSK2 blocked IAV-induced phosphorylation of double-stranded RNA-activated protein kinase (PKR), one of 4 known kinases (PKR, HRI, PERK and GCN2) that phosphorylate the translation-initiation factor elF2 during stress responses resulting in inhibition of cap-dependent translation of cellular and viral proteins [61,111].
PKR activation by influenza virus infections is well established and the virus has evolved multiple mechanisms to suppress PKR activation. Furthermore, IAV-dependent stimulation of NF-κB and IFN-β was impaired by RSK2 inhibition, suggesting an effect on the cellular antiviral response [61].
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Back to Top
1. Introduction
Influenza A (IAV) and B (IBV) viruses are important causes of upper respiratory tract infections [1]. IAV can cause severe acute respiratory disease with an attack rate of 5–10% in adults and 20–30% in children annually [2,3].
The significant public health burden caused by IAV infections is
exemplified by the annual fatal cases globally, which number
290,000–650,000 [4]. Most at risk are children and the elderly, accounting for ~90% of case fatalities and/or complications [5,6].
Occasionally, novel antigenically distinct influenza A viruses emerge
that may cause pandemic outbreaks as has occurred in 1918, 1957, 1968
and 2009. Unlike IAV, IBV viruses do not continuously circulate in
animals and are therefore less likely to be associated with zoonotic
transmission or pandemics [7]. However, they do co-circulate with IAV and can be significant contributors of influenza-related morbidity and mortality [7,8,9].
Vaccination is the preferred intervention against influenza viruses and
helps to limit the impact influenza outbreaks may have. In addition,
antiviral drugs are available for the treatment of influenza virus
infections. The surface glycoprotein hemagglutinin (HA) is the major
target for the induction of virus-neutralizing antibodies by
vaccination. Currently available antiviral drugs against influenza
inhibit the enzymatic activity of the viral neuraminidase (neuraminidase
inhibitors (NAIs)), inhibit the viral M2 ion channels or inhibit viral
RNA transcription by targeting components of the polymerase complex; all
of which ultimately result in inhibition of virus replication [10,11].
The use of antiviral drugs may lead to the emergence of strains that
have become resistant to these drugs by the accumulation of mutations
greatly reducing their efficacy [12].
This, of course, is an important drawback and there is a need for
treatment options that do not suffer from the emergence of resistant
strains***. Drugs that target host factors critical for virus replication
may therefore be an attractive alternative.
IAVs are members of the Orthomyxoviridae
family and have a negative-sense single-stranded RNA genome. Attachment
of IAV to cell-surface receptors, containing either α2,3- or
α2,6-linked sialic acid residues, initiates signaling cascades that
facilitate internalization of the virus via receptor mediated
endocytosis. During endosomal trafficking, pH-dependent fusion of viral
and endosomal membranes leads to release of viral ribonucleoproteins
(vRNPs) into the cellular cytoplasm where released vRNPs are shuttled to
the nucleus for replication and transcription of viral RNA; all of
which require host cell machinery [13].
These early events ultimately trigger multiple anti- and pro-viral
pathways utilized, suppressed or evaded by IAV. The robust production of
pro-inflammatory cytokines and chemokines observed during severe IAV
infections is often referred to as a “cytokine-storm” (reviewed in [14]).
This dysregulated immune response is associated with severe influenza
induced pneumonia that can be fatal, especially in susceptible
populations including children, older adults and the immunocompromised [15].
In
contrast to IAV, IBV is understudied, with only a few studies
addressing the role of host factors, and specifically kinases, and their
role during IBV infections. A better understanding of the viral and
cellular processes, mechanisms and interactions is required to develop
new treatment options [7].
Considering the overlap of IAV- and IBV-utilized kinases and their
related cellular signaling cascades to prime viral replication, defining
these pathways is likely to help in developing comprehensive
***host-targeted antivirals against IAV and IBV.
Kinases
link a myriad of external stimuli with downstream effectors through
phosphorylation of proteins and/or lipids. So far, more than 500 kinases
have been identified in the human kinome [16].
These kinases are typically categorized based on their phosphorylation
substrate: tyrosine, serine/threonine or lipids; as well as kinases that
have dual-specificity. Target residues (Tyr, Ser, Thr) are generally
within well-defined consensus sequence motifs recognized by a given
kinase [17,18,19].
Interestingly, the distribution of protein phosphorylation in
eukaryotic cells is distributed at a ratio of ~1000:100:1
(serine:threonine:tyrosine) [20]. Phosphorylation can alter activity and subcellular localization, as well as biomolecular interactions [21].
In addition, phosphorylation can promote scaffolding activities of
proteins that enhance, inhibit and modulate the substrates interaction
with other cellular components [22].
Aberrant kinase activity is typically associated with several
pathologies including cancer, diabetes or neurodegenerative diseases,
which has led to the development and investigation of several kinase
inhibitors for clinical use [23,24,25,26]. However, as of 2018,*** only 30 small-molecule kinase inhibitors (SMKIs) have gained FDA-approval for clinical use [26,27].
No SMKIs are currently under clinical trial investigation against
influenza virus infections.*** IAV does not encode a kinase and is
therefore dependent on cellular kinases to directly or indirectly,
regulate phosphorylation-dependent processes including viral entry and
uncoating, viral RNA and protein synthesis, protein relocation and
release of viral particles [28,29,30,31].
In addition, several studies have illustrated the importance of
IAV-protein phosphorylation in regulating viral replication and
evasion/suppression of innate immune signaling cascades that control
expression of pro inflammatory chemokines and cytokines response [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47]. Moreover, RNAi screenings continue to add to the list of host factors that impact IAV replication [48].
Therefore,
a better understanding is required of how influenza viruses utilize
these critical host factors and how these factors regulate
species-specific host adaption and pathogenesis of influenza viruses.
This review aims to discuss current knowledge of the role cellular
kinases play during in vitro and in vivo influenza virus infections ***as
potential antiviral targets (Table 1) (Figure 1).
Given the current state of knowledge, this review will be largely
focused on IAV studies; however, IBV studies will be highlighted when
possible.
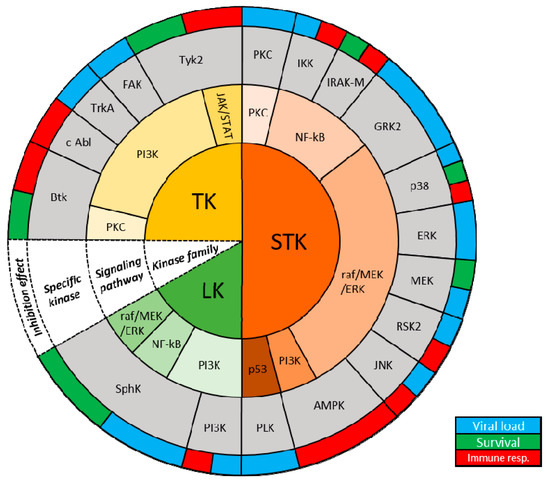
Figure 1.
Host kinases and known roles during IAV infections. Schematic organizing
host kinases based on kinase family, signaling pathway involved,
specific kinase and effect of inhibition (from innermost to outermost
ring; white cone).
Table 1.
Overview of cellular kinases and their role in different stages of IAV replication.
2. Phosphorylation of Influenza Virus Proteins
Phosphorylation of IAV and IBV has been reported with some conservation across influenza virus species [34].
IAV protein-phosphorylation regulates different stages of the viral
cycle by either promoting replication or evading/suppressing the innate
immune response [32,33,34,35,36,69].
Moreover, ***0treatment with kinase inhibitors affects influenza A virus
RNA and protein synthesis, shuttling of viral proteins between the
cytoplasm and nucleus, and virion release [28,29,30,31,80]. The nonstructural protein NS1 is a multifunctional immune modulator that counteracts host defenses [81,82].
NS1 phosphorylation at T215, S42 and S48 is thought to regulate the
dsRNA binding capacity of NS1, which promotes evasion of the innate
immune response [33,83].
Akt, an effector kinase of both the PI3K and ERK pathways, is
responsible for T215 phosphorylation, which consequently results in
viral entry and genome replication suppression following Akt inhibitor
treatment [84,85]. Additionally, mutation of S42 eliminates the interaction of NS1 with dsRNA and attenuates viral replication [33,84].
T132 phosphorylation of the M1 protein controls its nuclear import,
which is critical for viral replication. The Janus kinase 2 (JAK2)
inhibitor AG490 prevents nuclear import of M1, suggesting that JAK2
might be responsible for M1 T132 phosphorylation [34,35].
Inhibition of IAV nucleoprotein (NP) phosphorylation leads to its
nuclear retention that is largely regulated by several phosphorylation
sites, including S9, Y10, S165 and Y296. Mutation of these sites results
in decreased viral replication in vitro and in vivo largely through
disruption of interactions with cellular importin-α and chromosomal
maintenance 1 proteins [34,36,86].
3. Tyrosine Kinases (TK)
Tyrosine
kinases (TK) are a subgroub of ~90 kinases within the human kinome that
phosphorylate tyrosine amino acid residues; this can lead to
conformational changes in a given protein or even serve as a scaffolding
site to facilitate protein–protein interactions. TKs are further
classified into receptor tyrosine kinases (RTK) and non-receptor
tyrosine kinases (non-RTK). Non-RTK act as intracellular signal
transducers, mediating the signaling of cell-surface receptors for
cytokines, growth factors and other ligands [16]. Several phosphorylation sites (S, T and Y) in IAV and IBV proteins have been identified [34]. Based on additional sequence analysis of about 50,000 strains (www.fludb.org),
we identified highly conserved tyrosine residues in replication complex
proteins (PA, PB1, PB2) and NP proteins of all IAV subtypes. This level
of conservation suggests an evolutionary importance that might be
exploited in understanding conserved functions and developing broadly
active therapeutics targeting TK. Interestingly, while many of these
phosphorylation sites have been previously reported and their importance
demonstrated or inferred, the kinases that carry out their
phosphorylation have yet to be experimentally validated.
Nerve
growth factor receptor (TrkA) is a receptor tyrosine kinase that was
shown to play a role in IAV viral RNA synthesis, vRNP nuclear export and
virion release. In vitro inhibition of TrkA has been shown to diminish
IAV RNA (vRNA, mRNA and cRNA) synthesis independently of NFκB signaling [29].
Interestingly, this reduction in RNA synthesis was largely due to
direct inhibition of CRM1-mediated export and subsequent nuclear
retention of IAV RNPs [29].
In addition, TrkA inhibition leads to reduced activation of the lipid
biosynthesis enzyme, farnesyl diphosphate synthase (FPPS), which is
known to modulate virion budding [28]. However, the exact mechanism of TrkA-mediated FPPS regulation remains undefined.
Focal
adhesion kinase (FAK) is a non-RTK and a component of focal adhesions
that tether the actin cytoskeleton to the extracellular matrix. We
previously showed that FAK links phosphatidylinositol-3 kinase (PI3K)
activation and cytoskeletal reorganization required for endosomal
trafficking during IAV entry [40]. Furthermore, FAK positively regulates IAV replication and polymerase activity of different IAV strains/subtypes [39,40]. Others have also reported roles for FAK during other viral infections [87,88,89,90,91,92]. FAK can modulate the cellular immune response by regulating various functions of T cells, B cells and macrophages [93,94,95].
Consistent with this, we have observed FAK dependent regulation of
innate immune responses during severe IAV infection in mice [96].
Abl1
(also known as Abelson murine leukemia viral oncogene homolog 1 or
c-Abl) is a cytoplasmic and nuclear non-RTK that phosphorylates CRK
(also known as p38 or proto-oncogene c-CRK), an adaptor protein required
for efficient replication of avian influenza viruses and subsequent
JNK-mediated apoptosis [97].
The viral nonstructural protein 1 (NS1) can disrupt Abl1-CRK
interactions via its Src homology binding motifs and thereby inhibit CRK
phosphorylation, ultimately resulting in IAV-subtype specific
pathogenicity as shown for the 1918 pandemic H1N1 virus [50,51].
Acute
respiratory distress syndrome (ARDS) and acute lung injury (ALI) due to
immune cell infiltration during severe IAV and ensuing secondary
bacterial infections can result in respiratory failure and are the main
causes of death in influenza-infected patients [98]. Bruton’s tyrosine kinase (Btk) can regulate TLR4-mediated activation in human neutrophils [99]. Interestingly, chemical inhibition of Btk can alleviate IAV induced ARDS symptoms in mice [49].
This effect is likely due to limiting damaging neutrophil activity and
production of pro-inflammatory chemokines and cytokines including TNF-α,
IL-1β, IL-6, KC, and MCP-1 during acute lung injury [49].
The
IFN receptor I and III-associated tyrosine kinase 2 (Tyk2) has emerged
as an important host factor targeting secondary bacterial infections.
The virally induced retention of IL-1β and GM-CSF diminishes the
bacterial-induced innate immune response that may allow the
establishment of secondary bacterial infection. Specific ex vivo
inhibition of Tyk2 resulted in impaired bacterial growth due to restored
IL-1β and GM-CSF levels in human alveolar tissues [52].
4. Serine/Threonine Kinases (STK)
Serine/Threonine
kinases (STKs) facilitate phosphorylation of protein at either serine
or threonine residues. STKs are central components of many cellular
signaling pathways including Raf/MEK/ERK, nuclear factor kappa-B (NF-κB)
and PKC [100,101,102].
The Ras-dependent Raf/MEK/ERK pathway is activated by almost all
cytokines and growth factors that bind to receptor tyrosine kinases,
cytokine receptors and G-protein coupled receptors [43]. Accordingly, the importance of Raf/MEK/ERK signaling for effective IAV replication has previously been demonstrated [31,60].
IAVs
utilize multiple mechanisms to hijack STKs to evade subsequent innate
immune responses.
c-Jun N-terminal kinases 1 and 2 (JNK1/JNK2) can regulate pro-inflammatory response induction and are upregulated by several IAV strains. IAV-mediated induction of JNK1/JNK2 activity triggers the Raf/MEK/ERK pathway, mediating production of chemokines and cytokines including tumor necrosis factor alpha (TNF-α), interferon β (IFN-β) and interleukin 6 (IL-6) [45]. Interestingly, recent studies suggest that JNK1-dependent phosphorylation of Bcl-2, a process normally observed as a starvation induced autophagy signal, is promoted by viral JNK1 activation resulting in virus-induced autophagy [46]. Chemical inhibition of JNK1/JNK2 resulted in reduced levels of pro-inflammatory cytokines in vivo [45]. Additionally, in vitro inhibition of JNK1/JNK2 results in impaired vRNA synthesis; however, the mechanism is yet to be defined [53].
c-Jun N-terminal kinases 1 and 2 (JNK1/JNK2) can regulate pro-inflammatory response induction and are upregulated by several IAV strains. IAV-mediated induction of JNK1/JNK2 activity triggers the Raf/MEK/ERK pathway, mediating production of chemokines and cytokines including tumor necrosis factor alpha (TNF-α), interferon β (IFN-β) and interleukin 6 (IL-6) [45]. Interestingly, recent studies suggest that JNK1-dependent phosphorylation of Bcl-2, a process normally observed as a starvation induced autophagy signal, is promoted by viral JNK1 activation resulting in virus-induced autophagy [46]. Chemical inhibition of JNK1/JNK2 resulted in reduced levels of pro-inflammatory cytokines in vivo [45]. Additionally, in vitro inhibition of JNK1/JNK2 results in impaired vRNA synthesis; however, the mechanism is yet to be defined [53].
As
a member of the mitogen-activated protein kinase (MAPK) family, p38 is
involved in several steps of the IAV infection cycle. IAV infected cells
expressing the antiapoptotic protein Bcl-2 show reduced viral titers
due to reduced vRNP export from the nucleus with no effect on virally
induced apoptosis. The antiapoptotic effect of Bcl-2 was reduced by
phosphorylation of its threonine 56 and serine 87 residues by
virus-induced p38 activity. Inhibition of p38 diminished viral
replication, vRNP export and apoptosis [103]. During early stages, TLR4 mediated viral activation of p38 MAPK is important for viral entry and replication [37,55].
Furthermore, in vivo inhibition of p38 MAPK directly limited excessive
cytokine expression through an IFN-dependent mechanism. This regulation
is mediated via phosphorylation of STAT1 and subsequent engagement of
the IFNβ promotor to regulate IFN-stimulated gene (ISG) expression [54].
Influenza virus induced perturbations of the intracellular redox
balance resulting in increased production of reactive oxygen species
(ROS) can also activate p38 [56,104].
Furthermore, NADPH oxidase 4 (NOX4)-regulated p38 and ERK activation
leads to increased ROS production during IAV infections in vitro [56,105,106].
Interestingly, mouse experiments suggest that the effects of Bcl-2 and
NOX4 may be gender dependent. Female mice exhibited reduced clinical
symptoms and viral titers; in contrast, higher IAV replication in male
mice correlated with higher expression of NOX4 and phosphorylation of
p38 [107].
The NF-κB signaling pathway is a central regulator of innate immune responses and the IkB kinase (IKK) is a direct target of the viral NS1 protein in counteracting the NF-κB mediated cellular antiviral response [63,108]. However, the majority of publications have shown that inhibition of NF-κB signaling diminishes viral replication in vitro and in vivo [64,65,66]; more specifically, lowered levels of pro-inflammatory factors, reduced caspase activity and therefore impaired caspase-mediated nuclear export of vRNP [62].
Interleukin 1 receptor-associated kinase-M (IRAK-M) is a NF-κB signaling related cellular kinase. During IAV induced pneumonia, IRAK-M acts a central regulator of inflammation of mucosal tissue in the respiratory tract. IRAK-M knockout mice challenged with IAV showed strongly increased lethality rate and decreased viral clearance [67].
The NF-κB signaling pathway is a central regulator of innate immune responses and the IkB kinase (IKK) is a direct target of the viral NS1 protein in counteracting the NF-κB mediated cellular antiviral response [63,108]. However, the majority of publications have shown that inhibition of NF-κB signaling diminishes viral replication in vitro and in vivo [64,65,66]; more specifically, lowered levels of pro-inflammatory factors, reduced caspase activity and therefore impaired caspase-mediated nuclear export of vRNP [62].
Interleukin 1 receptor-associated kinase-M (IRAK-M) is a NF-κB signaling related cellular kinase. During IAV induced pneumonia, IRAK-M acts a central regulator of inflammation of mucosal tissue in the respiratory tract. IRAK-M knockout mice challenged with IAV showed strongly increased lethality rate and decreased viral clearance [67].
The successful nuclear export of vRNP has been shown to depend directly on the viral activation of the Raf/MEK/ERK signaling pathway [31,109].
MAPK kinase (MEK) and extracellular signal-regulated kinase (ERK), belong to the group of classical mitogen-activated protein kinases (MAPK). MEKs have been shown to regulate IAV and IBV replication [31,109]. Several MEK inhibitors resulted in vRNP retention, reduced titers of progeny virus in vitro, and also improved mouse survival in vivo [57,58,59].
During early stages of IAV infection, ERK regulates the vacuolar H+-ATPase (V-ATPase) activity to mediate pH-dependent acidification of endosomes and subsequent fusion of the viral and endosomal membranes [41]. In vitro inhibition of ERK, a direct downstream mediator of MEK, impedes IAV vRNP nuclear import as well as export [41,60].
IAVs activation of Raf/MEK/ERK signaling also induces p90 ribosomal s6 kinases (RSK), which play an important role as downstream mediators of ERK signaling [61,110]. RSK2 is involved in regulation of cell growth and proliferation. RSK2 knockdown using shRNAs results in increased IAV and IBV replication and IAV polymerase activity [61]. Inhibition of RSK2 blocked IAV-induced phosphorylation of double-stranded RNA-activated protein kinase (PKR), one of 4 known kinases (PKR, HRI, PERK and GCN2) that phosphorylate the translation-initiation factor elF2 during stress responses resulting in inhibition of cap-dependent translation of cellular and viral proteins [61,111].
PKR activation by influenza virus infections is well established and the virus has evolved multiple mechanisms to suppress PKR activation. Furthermore, IAV-dependent stimulation of NF-κB and IFN-β was impaired by RSK2 inhibition, suggesting an effect on the cellular antiviral response [61].
In
addition to Raf/MEK/ERK kinases, the G protein-coupled receptor kinases
(GRKs) are also implicated in the induction of innate immunity
pathways. Recent phosphoproteomic studies identified GRK2 as an
important junction of cellular signaling pathways activated by IAV. In
vitro and in vivo inhibition of GRK2 resulted in decreased viral
replication [71],
while the exact function of GRK2 remains unclear.
Polo-like kinases (PLK) act as GRK nodes of cellular signaling and are crucial regulators of cell division and the cell cycle [112]. PLK1 has been described as acting as a pro-viral host factor for several viruses by phospho-regulating viral proteins [113,114]. A recent study shows that in vitro and ex vivo inhibition, as well as knockdown of PLK1, PLK3 and PLK4, results in impaired IAV replication [73].
Polo-like kinases (PLK) act as GRK nodes of cellular signaling and are crucial regulators of cell division and the cell cycle [112]. PLK1 has been described as acting as a pro-viral host factor for several viruses by phospho-regulating viral proteins [113,114]. A recent study shows that in vitro and ex vivo inhibition, as well as knockdown of PLK1, PLK3 and PLK4, results in impaired IAV replication [73].
Protein
kinase C (PKC) is a STK that regulates multiple cellular processes
including proliferation, differentiation, apoptosis and angiogenesis.
The functional versatility of PKC is dependent on its various isoforms
responding to different stimuli. The complexity of eleven different PKC
isoforms expressed in most tissues also limits understanding of their
function within different cell types [115].
Nevertheless, Kurokawa et al. showed almost 30 years ago that general
in vitro inhibition of PKC results in reduced viral protein synthesis [30].
More recent studies have further defined the function of PKC isoforms
and their involvement in IAV infections. Treatment of cells with
bisindolylmaleimide, a highly specific PKC inhibitor that has activity
against most PKC isoforms, reversibly inhibits virus entry by blocking
endosomal trafficking and virion uncoating of both IAV and IBV [80].
Phosphorylation of the viral proteins PB1 and NS1, important for
polymerase activity and efficient viral replication, has been shown to
be PKCα dependent in vitro [68] and for PB1 in vivo [69].
In PKCβII kinase-dead cells, IAV is retained in late endosomal
compartments, suggesting PKCβII as an important modulator of IAV entry [44].
PKCδ, interaction with the IAV polymerase subunit PB2, regulates NP
oligomerization and vRNP assembly, and ablation of PKCδ impaired
replication of the viral genome in vitro [70].
5. Lipid Kinases (LK)
Lipid
kinases are key mediators of intracellular signaling, central carbon
and lipid metabolism, apoptosis and cell proliferation through
phosphorylation of lipid residues. Several lipid kinases have been
implicated in several steps of IAV replication and in modulating
cellular antiviral responses [38,79,116,117,118].
One of the central lipid kinases is PI3K, which phosphorylates inositol phospholipids [119]. PI3K and its downstream effectors, Akt and mammalian target of rapamycin (mTOR), form a key signaling nexus that regulates cell differentiation, translation and metabolism [120].
Furthermore, it is involved in cross-interaction with other cellular signaling pathways including Raf/MEK/ERK and NF-κB pathways [121].
Early and late PI3K during IAV infections are key events required for IAV replication with distinct outcomes at different times of infection [38].
Early PI3K activity is triggered by viral attachment and mediates IAV entry [75].
Later during the infection, IAV NS1 suppresses PI3K activity via direct interactions with the p85 regulatory subunit. These interactions ultimately prevent AKT-mediated apoptosis, IRF-3 innate immune responses, vRNA synthesis and nuclear vRNP export
[38,74,75,76,77,122,123]. It should be noted that IBV only minimally induces later PI3K activation or apoptosis. Furthermore, in contrast to IAV NS1, IBV NS1 is dispensable for the antiapoptotic effects of PI3K activation suggesting IBV has developed NS1-independent mechanisms to suppress apoptosis [116,124].
One of the central lipid kinases is PI3K, which phosphorylates inositol phospholipids [119]. PI3K and its downstream effectors, Akt and mammalian target of rapamycin (mTOR), form a key signaling nexus that regulates cell differentiation, translation and metabolism [120].
Furthermore, it is involved in cross-interaction with other cellular signaling pathways including Raf/MEK/ERK and NF-κB pathways [121].
Early and late PI3K during IAV infections are key events required for IAV replication with distinct outcomes at different times of infection [38].
Early PI3K activity is triggered by viral attachment and mediates IAV entry [75].
Later during the infection, IAV NS1 suppresses PI3K activity via direct interactions with the p85 regulatory subunit. These interactions ultimately prevent AKT-mediated apoptosis, IRF-3 innate immune responses, vRNA synthesis and nuclear vRNP export
[38,74,75,76,77,122,123]. It should be noted that IBV only minimally induces later PI3K activation or apoptosis. Furthermore, in contrast to IAV NS1, IBV NS1 is dispensable for the antiapoptotic effects of PI3K activation suggesting IBV has developed NS1-independent mechanisms to suppress apoptosis [116,124].
Sphingosin
kinases (SphK1 and SphK2) are lipid kinases that control conversion of
sphingosine to bioactive lipid sphingosine 1-phosphate (S1P) [125], a known modulator of Raf/MEK/ERK, NF-κB and PI3K/AKT/mTOR signaling pathways and regulator of apoptosis [126].
IAV upregulates SphK in in vitro infected cells influencing cellular
signaling and promoting efficient influenza virus replication [78,79].
Chemical inhibition of SphK1 results in reduced vRNA synthesis via suppression of NF-κB activity and reduced vRNP nuclear export due to impaired activation of ERK and AKT [78]. SphK2 knockdown has also been shown to reduce IAV replication in vitro. Moreover, in vivo inhibition of SphK1 and SphK2 resulted in prolonged survival of mice challenged with IAV [79].
Chemical inhibition of SphK1 results in reduced vRNA synthesis via suppression of NF-κB activity and reduced vRNP nuclear export due to impaired activation of ERK and AKT [78]. SphK2 knockdown has also been shown to reduce IAV replication in vitro. Moreover, in vivo inhibition of SphK1 and SphK2 resulted in prolonged survival of mice challenged with IAV [79].
6. Linking Metabolism and Innate Immunity
Like
many pathologic conditions, IAV infection alters the metabolic
landscape and most of these alterations are mediated by kinases
resulting in direct or indirect effect on IAV replication, infection
kinetics and pathogenicity. Consistently, the majority of host-cell
alterations following IAV infections are in metabolic pathways [127].
Virus regulated kinase activity can have a major influence on cellular
metabolism.
AMP-activated protein kinase (AMPK) is a major sensor and regulatory master switch of carbohydrate metabolism, and is directly involved in insulin signaling and lipid metabolism. It links central carbon metabolism and glucose availability with the host innate immune response [128,129,130,131].
AMPK activity is modulated by intracellular calcium levels and this activity can regulate the stimulator of interferon genes (STING) through UNC-51-like kinase 1 (ULK1) activation. STING serves as a crucial factor of the innate immune response and an essential mediator for recognition of intracellular bacterial and viral pathogens. STING-dependent IFNβ induction is regulated by the calcium-dependent membrane potential of mitochondrial membranes.
In vitro inhibition of AMPK resulted in reduced TNF-α and IFN-β secretion after activation with the STING ligand 5,6-dimethyl xanthone-4 acetic acid (DMXAA) [132,133,134].
AMPK phosphorylation of multiple sites of ULK1 leads to its dissociation from AMPK and subsequent activation. ULK1 activity promotes phosphatidylinositol-3-phosphate (PI3P) synthesis that contributes to autophagosome formation in addition to JNK1 induced Bcl-2 dependent autophagy during IAV infection [46,135,136,137].
AMP-activated protein kinase (AMPK) is a major sensor and regulatory master switch of carbohydrate metabolism, and is directly involved in insulin signaling and lipid metabolism. It links central carbon metabolism and glucose availability with the host innate immune response [128,129,130,131].
AMPK activity is modulated by intracellular calcium levels and this activity can regulate the stimulator of interferon genes (STING) through UNC-51-like kinase 1 (ULK1) activation. STING serves as a crucial factor of the innate immune response and an essential mediator for recognition of intracellular bacterial and viral pathogens. STING-dependent IFNβ induction is regulated by the calcium-dependent membrane potential of mitochondrial membranes.
In vitro inhibition of AMPK resulted in reduced TNF-α and IFN-β secretion after activation with the STING ligand 5,6-dimethyl xanthone-4 acetic acid (DMXAA) [132,133,134].
AMPK phosphorylation of multiple sites of ULK1 leads to its dissociation from AMPK and subsequent activation. ULK1 activity promotes phosphatidylinositol-3-phosphate (PI3P) synthesis that contributes to autophagosome formation in addition to JNK1 induced Bcl-2 dependent autophagy during IAV infection [46,135,136,137].
Although
ER stress triggers translational shut-down through the PKR-like ER
kinase (PERK), virally induced metabolic and ER stress in the context of
an obese mouse model activates PKR [138]. This activation reduces cellular and viral translation and activates JNK1 and other inflammatory kinases in response [138].
Together, PKR and nutrient deprivation-dependent JNK1 activities lead
to the subsequent activation of apoptosis signal regulating kinase 1
(ASK1) [139].
Integration of AMPK and JNK with other Raf/MEK/ERK related kinases allows engagement of metabolic processes via immune response components including NF-κB, PI3K/AKT/mTOR and PKC pathways [117,118,140,141,142].
Accordingly, the NF-κB regulating kinase, IKK, has recently been linked to glycolysis [143,144].
In addition, IKK- and PKC-dependent serine phosphorylation of the insulin receptor, inhibits insulin signaling and directly regulates cellular lipid metabolism [145,146].
Furthermore, PKC has been described to be involved in fatty acid fate regulation, auto-stimulating kinase activity [147]. PI3K/AKT/mTOR signaling mediates its effects upstream and downstream of NF-κB, Raf/MEK/ERK and PKC pathways to regulate lipogenesis and lipid metabolism [121,131,148,149].
Recent studies suggest that inhibition of Btk leads to metabolic stress through suppression of PI3K/AKT/mTOR signaling [150], highlighting the link between metabolism and innate immunity.
Interestingly, using a PI3K/mTOR inhibitor to disrupt glucose metabolism in vitro results in reduced virus production independently of genome replication and most likely drives lipid membrane depletion due to viral budding [127].
It is important to note that influenza virus-induced kinase activity does not only serve to evade the immune response but can also promote a pro-viral metabolic environment and responses.
Integration of AMPK and JNK with other Raf/MEK/ERK related kinases allows engagement of metabolic processes via immune response components including NF-κB, PI3K/AKT/mTOR and PKC pathways [117,118,140,141,142].
Accordingly, the NF-κB regulating kinase, IKK, has recently been linked to glycolysis [143,144].
In addition, IKK- and PKC-dependent serine phosphorylation of the insulin receptor, inhibits insulin signaling and directly regulates cellular lipid metabolism [145,146].
Furthermore, PKC has been described to be involved in fatty acid fate regulation, auto-stimulating kinase activity [147]. PI3K/AKT/mTOR signaling mediates its effects upstream and downstream of NF-κB, Raf/MEK/ERK and PKC pathways to regulate lipogenesis and lipid metabolism [121,131,148,149].
Recent studies suggest that inhibition of Btk leads to metabolic stress through suppression of PI3K/AKT/mTOR signaling [150], highlighting the link between metabolism and innate immunity.
Interestingly, using a PI3K/mTOR inhibitor to disrupt glucose metabolism in vitro results in reduced virus production independently of genome replication and most likely drives lipid membrane depletion due to viral budding [127].
It is important to note that influenza virus-induced kinase activity does not only serve to evade the immune response but can also promote a pro-viral metabolic environment and responses.
7. Perspectives and Future Directions
The
continued threat of severe and potentially lethal influenza A virus
outbreaks is highlighted by rapid viral evolution, emergence of novel
subtypes and antiviral-resistant strains and limited vaccine efficacy.
Developing virus-directed antivirals is akin to hitting a moving target.
Therefore, approaches that largely mitigate the potential for
drug-resistance while being effective against multiple IAV subtypes and
strains is highly desirable. Therapies that target host cell factors
meet these criteria and are more likely to avoid exuberant immune
responses that are likely to reduce disease severity and improve patient
outcome.
***Kinases are ideal candidates for
host-directed antiviral therapies by linking critical cellular processes
utilized by most viruses. Moreover, their importance in pathologic
conditions such as cancer has led to the development of ***small-molecule
inhibitors and repurposing these clinically approved drugs to treat
severe infectious diseases like influenza, should be exploited.
Several
reports have recently highlighted critical roles for the focal adhesion
kinase (FAK) pathway during infection by several viruses [87,88,89,90,91].
FAK is not only critical for embryonic development and expression of
several cellular proteins, it also links integrins with actin
reorganization and receptor endocytosis [151,152,153,154].
Given its role in several cancers and the unique structure of its
kinase domain, FAK is an attractive target of anti-cancer therapies and
several FAK inhibitors are under investigation for clinical use [155].
The
FAK pathway has recently emerged as a nexus point engaging antiviral
innate immune and inflammatory pathways. Accordingly, FAK is also a
component of the intracellular RIG-I-like receptor antiviral pathway
where it provides a link between perturbations of the cell surface
receptor during viral entry and cytosolic innate immune sensors [156]. FAK modulates the cellular immune response by regulating T cells, B cells and macrophage functions [93,94,95]. FAK was also recently reported to directly phosphorylate IKKα thereby regulating canonical and non-canonical NF-κB pathways [157].
Although
SMKIs have been met with often-warranted criticism, this has stemmed
from a misconception in clinical literature and inaccurate distinction
between in vitro/in vivo substrate (target) specificity and
cell-population specificity in vivo of these SMKIs [158,159,160,161].
Because tyrosine kinases share conserved sequences in their ATP binding sites, ATP analogs have an increased likelihood of “off-target” effects on other kinases [162]. Therefore, new small molecule inhibitors designed to avoid this problem directly interfere with FAK autophosphorylation by binding to Y397 instead of blocking ATP binding. One such compound is FAK Inhibitor I (also known as Compound 14 or Y15) which has been validated as a selective FAK inhibitor [163,164,165]. We found that Y15-treatment of various cells, or expression of kinase-dead FAK mutant (FAK-KD), provided the first evidence that FAK is activated by IAV attachment and that FAK kinase activity is critical for efficient endosomal virus trafficking [40].
We also reported that inhibitor-treatment or FAK-KD expression reduced polymerase activity of multiple IAV subtypes including highly pathogenic H5N1 and H7N9. Importantly, we observed FAK interactions with the viral NP [39]; however, the significance of this interaction is still under investigation.
Defactinib is an FDA approved FAK inhibitor that has dual activity against FAK and the related kinase Pyk2 and is therefore expected to have different effects than Y15 due to differences in specificities. Our published data utilizing Y15 clearly indicates a FAK specific role in IAV replication. However, given that Pyk2 has overlapping roles in immune cell development and functions [93,94,95], it is possible that inhibiting both kinases will have alternative outcomes. While this might first be viewed as a cause for concern, it provides the opportunity to potentially fine tune treatments where either FAK or Pyk2 or both can be inhibited depending on the timing of treatment (early vs late in infection).
Because tyrosine kinases share conserved sequences in their ATP binding sites, ATP analogs have an increased likelihood of “off-target” effects on other kinases [162]. Therefore, new small molecule inhibitors designed to avoid this problem directly interfere with FAK autophosphorylation by binding to Y397 instead of blocking ATP binding. One such compound is FAK Inhibitor I (also known as Compound 14 or Y15) which has been validated as a selective FAK inhibitor [163,164,165]. We found that Y15-treatment of various cells, or expression of kinase-dead FAK mutant (FAK-KD), provided the first evidence that FAK is activated by IAV attachment and that FAK kinase activity is critical for efficient endosomal virus trafficking [40].
We also reported that inhibitor-treatment or FAK-KD expression reduced polymerase activity of multiple IAV subtypes including highly pathogenic H5N1 and H7N9. Importantly, we observed FAK interactions with the viral NP [39]; however, the significance of this interaction is still under investigation.
Defactinib is an FDA approved FAK inhibitor that has dual activity against FAK and the related kinase Pyk2 and is therefore expected to have different effects than Y15 due to differences in specificities. Our published data utilizing Y15 clearly indicates a FAK specific role in IAV replication. However, given that Pyk2 has overlapping roles in immune cell development and functions [93,94,95], it is possible that inhibiting both kinases will have alternative outcomes. While this might first be viewed as a cause for concern, it provides the opportunity to potentially fine tune treatments where either FAK or Pyk2 or both can be inhibited depending on the timing of treatment (early vs late in infection).
Investigating
***repurposed cancer drugs for their antiviral properties and their
potential immunomodulatory effects during infection will improve our
understanding of the role of the respective kinases in the pathogenesis
of IAV infections and may lead to the development of novel intervention
strategies.
Further research on the role of host kinases in virus-induced metabolic changes is warranted and will likely open-up additional avenues of basic and translational research.
Further research on the role of host kinases in virus-induced metabolic changes is warranted and will likely open-up additional avenues of basic and translational research.
Author Contributions
R.M.,
G.F.R. and H.E. conceptualized and composed the manuscript. G.F.R. and
H.E. oversaw all aspects of the manuscript preparation.
Funding
This
research and the APC was funded by the Alexander von Humboldt
Foundation in the framework of the Alexander von Humboldt Professorship
endowed by the German Federal Ministry of Education and Research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza a viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [PubMed]
- Moore, H.C.; Lehmann, D.; de Klerk, N.; Smith, D.W.; Richmond, P.C.; Keil, A.D.; Blyth, C.C. How accurate are international classification of diseases-10 diagnosis codes in detecting influenza and pertussis hospitalizations in children? J. Pediatr. Infect. Dis. Soc. 2014, 3, 255–260. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Influenza (Seasonal) Fact Sheet. Available online: http://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 12 November 2018).
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef] [PubMed]
- van de Sandt, C.E.; Bodewes, R.; Rimmelzwaan, G.F.; de Vries, R.D. Influenza b viruses: Not to be discounted. Future Microbiol. 2015, 10, 1447–1465. [Google Scholar] [CrossRef] [PubMed]
- Paul Glezen, W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza b: A structured literature review. Am. J. Public Health 2013, 103, e43–e51. [Google Scholar] [CrossRef] [PubMed]
- Koutsakos, M.; Nguyen, T.H.; Barclay, W.S.; Kedzierska, K. Knowns and unknowns of influenza b viruses. Future Microbiol. 2016, 11, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef]
- Ison, M.G. Antiviral treatments. Clin. Chest Med. 2017, 38, 139–153. [Google Scholar] [CrossRef]
- Carrat, F.; Flahault, A. Influenza vaccine: The challenge of antigenic drift. Vaccine 2007, 25, 6852–6862. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza a virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.N. Structural basis for substrate recognition and control in protein kinases. In Data Mining in Structural Biology; Springer: Berlin/Heidelberg, Germany, 2001; pp. 47–69. [Google Scholar]
- Jacob, T.; Van den Broeke, C.; Favoreel, H.W. Viral serine/threonine protein kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar]
- Raggiaschi, R.; Gotta, S.; Terstappen, G.C. Phosphoproteome analysis. Biosci. Rep. 2005, 25, 33–44. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. Erk and p38 mapk-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Frojdo, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta 2009, 1792, 83–92. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flight, M.H. Neurodegenerative diseases: New kinase targets for alzheimer’s disease. Nat. Rev. Drug Discov. 2013, 12, 739. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Hijazi, M.; Britton, D.; Cutillas, P.R. Impact of phosphoproteomics in the translation of kinase-targeted therapies. Proteomics 2017, 17, 1600235. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of fda-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef]
- Kumar, N.; Liang, Y.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication. J. Virol. 2011, 85, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Sharma, N.R.; Ly, H.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors that block replication of influenza a and other viruses. Antimicrob. Agents Chemother. 2011, 55, 5553–5559. [Google Scholar] [CrossRef]
- Kurokawa, M.; Ochiai, H.; Nakajima, K.; Niwayama, S. Inhibitory effect of protein kinase c inhibitor on the replication of influenza type a virus. J. Gen. Virol. 1990, 71, 2149–2155. [Google Scholar] [CrossRef]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the raf/mek/erk signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef]
- Arrese, M.; Portela, A. Serine 3 is critical for phosphorylation at the n-terminal end of the nucleoprotein of influenza virus a/victoria/3/75. J. Virol. 1996, 70, 3385–3391. [Google Scholar]
- Hsiang, T.-Y.; Zhou, L.; Krug, R.M. Roles of the phosphorylation of specific serines and threonines in the ns1 protein of human influenza a viruses. J. Virol. 2012, 86, 10370–10376. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Denham, E.M.; Thomas, B.; Trudgian, D.C.; Hester, S.S.; Ridlova, G.; York, A.; Turrell, L.; Fodor, E. Mapping the phosphoproteome of influenza a and b viruses by mass spectrometry. PLoS Pathog. 2012, 8, e1002993. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Z.; Bi, Y.; Sun, L.; Liu, X.; Liu, W. Tyrosine 132 phosphorylation of influenza a virus m1 protein is crucial for virus replication by controlling the nuclear import of m1. J. Virol. 2013, 87, 6182–6191. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, J.; Wang, S.; Cao, S.; Jiang, J.; Chen, C.; Ding, C.; Qin, C.; Ye, X.; Gao, G.F.; et al. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza a virus nucleoprotein. J. Virol. 2015, 89, 5822–5834. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Heo, J.; Yi, C.M.; Ban, J.; Lee, N.J.; Lee, N.R.; Kim, S.W.; Kim, N.J.; Inn, K.S. A novel p38 mitogen activated protein kinase (mapk) specific inhibitor suppresses respiratory syncytial virus and influenza a virus replication by inhibiting virus-induced p38 mapk activation. Biochem. Biophys. Res. Commun. 2016, 477, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C. From virus entry to release: The diverse functions of pi3k during rna virus infections. Future Virol. 2011, 6, 1225–1239. [Google Scholar] [CrossRef]
- Elbahesh, H.; Bergmann, S.; Russell, C.J. Focal adhesion kinase (fak) regulates polymerase activity of multiple influenza a virus subtypes. Virology 2016, 499, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Elbahesh, H.; Cline, T.; Baranovich, T.; Govorkova, E.A.; Schultz-Cherry, S.; Russell, C.J. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza a viruses. J. Virol. 2014, 88, 6714–6728. [Google Scholar] [CrossRef]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza a virus-induced early activation of erk and pi3k mediates v-atpase-dependent intracellular ph change required for fusion. Cell. Microbiol. 2011, 13, 587–601. [Google Scholar] [CrossRef]
- Ohmichi, M.; Pang, L.; Ribon, V.; Gazit, A.; Levitzki, A.; Saltiel, A.R. The tyrosine kinase inhibitor tyrphostin blocks the cellular actions of nerve growth factor. Biochemistry 1993, 32, 4650–4658. [Google Scholar] [CrossRef]
- Planz, O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antivir. Res. 2013, 98, 457–468. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Brown, H.A.; Whittaker, G.R. Role of protein kinase c betaii in influenza virus entry via late endosomes. J. Virol. 2003, 77, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, S.; Hu, Y.; Li, D.; Cui, J.; Xue, J.; Zhang, G.; Khachigian, L.M.; Wong, J.; Sun, L.; et al. Regulatory roles of c-jun in h5n1 influenza virus replication and host inflammation. Biochim. Biophys. Acta 2014, 1842, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ruan, T.; Sheng, T.; Wang, J.; Sun, J.; Wang, J.; Prinz, R.A.; Peng, D.; Liu, X.; Xu, X. Role of c-jun terminal kinase (jnk) activation in influenza a virus-induced autophagy and replication. Virology 2018, 526, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S. Targeting cell signalling pathways to fight the flu: Towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009, 64, 1–4. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Davis, S.A.; Matthay, M.A.; Kurdowska, A.K. Inhibiting bruton’s tyrosine kinase rescues mice from lethal influenza-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L52–l58. [Google Scholar] [CrossRef] [PubMed]
- Hrincius, E.R.; Liedmann, S.; Anhlan, D.; Wolff, T.; Ludwig, S.; Ehrhardt, C. Avian influenza viruses inhibit the major cellular signalling integrator c-abl. Cell. Microbiol. 2014, 16, 1854–1874. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Liedmann, S.; Finkelstein, D.; Vogel, P.; Gansebom, S.; Ehrhardt, C.; Ludwig, S.; Hains, D.S.; Webby, R.; McCullers, J.A. Nonstructural protein 1 (ns1)-mediated inhibition of c-abl results in acute lung injury and priming for bacterial co-infections: Insights into 1918 h1n1 pandemic? J. Infect. Dis. 2015, 211, 1418–1428. [Google Scholar] [CrossRef]
- Berg, J.; Zscheppang, K.; Fatykhova, D.; Tonnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Eggeling, S.; Schimek, M.; et al. Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur. Respir. J. 2017, 50, 1601953. [Google Scholar] [CrossRef]
- Zhang, S.; Tian, H.; Cui, J.; Xiao, J.; Wang, M.; Hu, Y. The c-jun n-terminal kinase (jnk) is involved in h5n1 influenza a virus rna and protein synthesis. Arch. Virol. 2016, 161, 345–351. [Google Scholar] [CrossRef]
- Borgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal h5n1 infection. J. Biol Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.; Palamara, A. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by nox4-derived ros. Cell. Microbiol. 2014, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Droebner, K.; Pleschka, S.; Ludwig, S.; Planz, O. Antiviral activity of the mek-inhibitor u0126 against pandemic h1n1v and highly pathogenic avian influenza virus in vitro and in vivo. Antivir. Res. 2011, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Hartmayer, C.; Planz, O. Combination of mek inhibitors and oseltamivir leads to synergistic antiviral effects after influenza a virus infection in vitro. Antivir. Res. 2013, 98, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Haasbach, E.; Muller, C.; Ehrhardt, C.; Schreiber, A.; Pleschka, S.; Ludwig, S.; Planz, O. The mek-inhibitor ci-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antivir. Res. 2017, 142, 178–184. [Google Scholar] [CrossRef]
- Marjuki, H.; Alam, M.I.; Ehrhardt, C.; Wagner, R.; Planz, O.; Klenk, H.D.; Ludwig, S.; Pleschka, S. Membrane accumulation of influenza a virus hemagglutinin triggers nuclear export of the viral genome via protein kinase calpha-mediated activation of erk signaling. J. Biol. Chem. 2006, 281, 16707–16715. [Google Scholar] [CrossRef]
- Kakugawa, S.; Shimojima, M.; Goto, H.; Horimoto, T.; Oshimori, N.; Neumann, G.; Yamamoto, T.; Kawaoka, Y. Mitogen-activated protein kinase-activated kinase rsk2 plays a role in innate immune responses to influenza virus infection. J. Virol. 2009, 83, 2510–2517. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Ruckle, A.; Hrincius, E.R.; Haasbach, E.; Anhlan, D.; Ahmann, K.; Banning, C.; Reiling, S.J.; Kuhn, J.; Strobl, S.; et al. The nf-kappab inhibitor sc75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell. Microbiol. 2013, 15, 1198–1211. [Google Scholar] [CrossRef]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza a virus-encoded ns1 virulence factor protein inhibits innate immune response by targeting ikk. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef]
- Haasbach, E.; Reiling, S.J.; Ehrhardt, C.; Droebner, K.; Ruckle, A.; Hrincius, E.R.; Leban, J.; Strobl, S.; Vitt, D.; Ludwig, S.; et al. The nf-kappab inhibitor sc75741 protects mice against highly pathogenic avian influenza a virus. Antivir. Res. 2013, 99, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active nf-kappab signalling is a prerequisite for influenza virus infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. Nf-kappab-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (trail) and fas/fasl is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Kohno, S.; Newstead, M.W.; Zeng, X.; Bhan, U.; Lukacs, N.W.; Kunkel, S.L.; Standiford, T.J. Critical role of il-1 receptor-associated kinase-m in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J. Immunol. 2010, 184, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, S.; Auerochs, S.; Grone, M.; Marschall, M. Influenza a virus proteins pb1 and ns1 are subject to functionally important phosphorylation by protein kinase C. J. Gen. Virol. 2009, 90, 1392–1397. [Google Scholar] [CrossRef] [PubMed]
- Mitzner, D.; Dudek, S.E.; Studtrucker, N.; Anhlan, D.; Mazur, I.; Wissing, J.; Jansch, L.; Wixler, L.; Bruns, K.; Sharma, A.; et al. Phosphorylation of the influenza a virus protein pb1-f2 by pkc is crucial for apoptosis promoting functions in monocytes. Cell. Microbiol. 2009, 11, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Dawson, A.R.; Potts, G.K.; Freiberger, E.C.; Baker, S.F.; Moser, L.A.; Bernard, K.A.; Coon, J.J.; Mehle, A. Influenza virus recruits host protein kinase c to control assembly and activity of its replication machinery. Elife 2017, 6, e26910. [Google Scholar] [CrossRef] [PubMed]
- Yanguez, E.; Hunziker, A.; Dobay, M.P.; Yildiz, S.; Schading, S.; Elshina, E.; Karakus, U.; Gehrig, P.; Grossmann, J.; Dijkman, R.; et al. Phosphoproteomic-based kinase profiling early in influenza virus infection identifies grk2 as antiviral drug target. Nat. Commun. 2018, 9, 3679. [Google Scholar] [CrossRef]
- Moseley, C.E.; Webster, R.G.; Aldridge, J.R. Peroxisome proliferator-activated receptor and amp-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir. Viruses 2010, 4, 307–311. [Google Scholar] [CrossRef]
- Pohl, M.O.; von Recum-Knepper, J.; Rodriguez-Frandsen, A.; Lanz, C.; Yanguez, E.; Soonthornvacharin, S.; Wolff, T.; Chanda, S.K.; Stertz, S. Identification of polo-like kinases as potential novel drug targets for influenza a virus. Sci. Rep. 2017, 7, 8629. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: Influenza viruses and the pi3k/akt signalling pathway. Cell. Microbiol. 2009, 11, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Marjuki, H.; Wolff, T.; Nurnberg, B.; Planz, O.; Pleschka, S.; Ludwig, S. Bivalent role of the phosphatidylinositol-3-kinase (pi3k) during influenza virus infection and host cell defence. Cell. Microbiol. 2006, 8, 1336–1348. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza a virus ns1 protein activates the pi3k/akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/akt pathway on influenza a virus propagation. J. Gen. Virol. 2007, 88, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Pritzl, C.J.; Vijayan, M.; Bomb, K.; McClain, M.E.; Alexander, S.; Hahm, B. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral rna synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PLoS ONE 2013, 8, e75005. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Seo, Y.J.; Studstill, C.J.; Vijayan, M.; Wolf, J.J.; Hahm, B. Transient inhibition of sphingosine kinases confers protection to influenza a virus infected mice. Antivir. Res. 2018, 158, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Root, C.N.; Wills, E.G.; McNair, L.L.; Whittaker, G.R. Entry of influenza viruses into cells is inhibited by a highly specific protein kinase c inhibitor. J. Gen. Virol. 2000, 81, 2697–2705. [Google Scholar] [CrossRef]
- Ayllon, J.; Garcia-Sastre, A. The ns1 protein: A multitasking virulence factor. Curr. Top Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar]
- Krug, R.M. Functions of the influenza a virus ns1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional ns1 protein of influenza a viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. Cdk/erk-mediated phosphorylation of the human influenza a virus ns1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Suizu, F.; Matsuda-Lennikov, M.; Edamura, T.; Bala, J.; Noguchi, M. Inhibition of akt kinase activity suppresses entry and replication of influenza virus. Biochem. Biophys. Res. Commun. 2014, 450, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; Di Primo, C.; Baudin, F.; Li, X.; Crepin, T.; Ruigrok, R.W. Monomeric nucleoprotein of influenza a virus. PLoS Pathog. 2013, 9, e1003275. [Google Scholar] [CrossRef] [PubMed]
- Cheshenko, N.; Liu, W.; Satlin, L.M.; Herold, B.C. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 2005, 280, 31116–31125. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, P.M.; Keiser, N.W.; Yan, Z.; Lei-Butters, D.C.; Engelhardt, J.F. Directing integrin-linked endocytosis of recombinant aav enhances productive fak-dependent transduction. Mol. Ther. 2012, 20, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Sharma-Walia, N.; Streblow, D.N.; Naranatt, P.P.; Chandran, B. Focal adhesion kinase is critical for entry of kaposi’s sarcoma-associated herpesvirus into target cells. J. Virol. 2006, 80, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.; Schneider, R.J. Activation of focal adhesion kinase by hepatitis b virus hbx protein: Multiple functions in viral replication. J. Virol. 2006, 80, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, B.; Nikolic, J.; Larrous, F.; Bourhy, H.; Wirblich, C.; Lagaudriere-Gesbert, C.; Blondel, D. Focal adhesion kinase is involved in rabies virus infection through its interaction with viral phosphoprotein p. J. Virol. 2015, 89, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.; Wen, L.B.; Wang, R.; Hao, H.P.; Huan, C.C.; Wang, X.; Huang, L.; Miao, J.F.; Fan, H.J.; Mao, X. The involvement of fak-pi3k-akt-rac1 pathway in porcine reproductive and respiratory syndrome virus entry. Biochem. Biophys. Res. Commun. 2015, 458, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Connolly, S.F.; Reinl, E.L.; Houtman, J.C. Focal adhesion kinase negatively regulates lck function downstream of the t cell antigen receptor. J. Immunol. 2013, 191, 6208–6221. [Google Scholar] [CrossRef]
- St-Pierre, J.; Ostergaard, H.L. A role for the protein tyrosine phosphatase cd45 in macrophage adhesion through the regulation of paxillin degradation. PLoS ONE 2013, 8, e71531. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Wolfram, P.; Canty, K.; Harley, B.; Nombela-Arrieta, C.; Pivarnik, G.; Manis, J.; Beggs, H.E.; Silberstein, L.E. Focal adhesion kinase regulates the localization and retention of pro-b cells in bone marrow microenvironments. J. Immunol. 2013, 190, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Bergman, S.; Elbahesh, H. Targeting the proviral host kinase, FAK, limits in vivo and in vitro influenza A virus pathogenesis and NFkB-regulated pro-inflammatory responses. Virology 2019. under review. [Google Scholar]
- Hrincius, E.R.; Wixler, V.; Wolff, T.; Wagner, R.; Ludwig, S.; Ehrhardt, C. Crk adaptor protein expression is required for efficient replication of avian influenza a viruses and controls jnk-mediated apoptotic responses. Cell. Microbiol. 2010, 12, 831–843. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Krupa, A.; Fudala, R.; Florence, J.M.; Tucker, T.; Allen, T.C.; Standiford, T.J.; Luchowski, R.; Fol, M.; Rahman, M.; Gryczynski, Z.; et al. Bruton’s tyrosine kinase mediates fcgammariia/toll-like receptor-4 receptor crosstalk in human neutrophils. Am. J. Respir. Cell. Mol. Biol. 2013, 48, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T.J.A. Function and activation of nf-kappab in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Graves, J.; Campbell, J.; Krebs, E.J.A. Protein serine/threonine kinases of the mapk cascade. Ann. N. Y. Acad. Sci. 1995, 766, 320–343. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase c: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38mapk activity in cells infected with influenza a virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar] [CrossRef]
- Lucia, N.; Rossella, S.; Donatella, A.; Paola, C.; Ignacio, C.; Dolores, L.; Simona, A.; Anna Teresa, P.; Enrico, G. Intracellular redox signaling as therapeutic target for novel antiviral strategy. Curr. Pharm. Des. 2011, 17, 3898–3904. [Google Scholar]
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 mediates the expression of plasminogen activator inhibitor-1 via p38 mapk pathway in cultured human endothelial cells. Thromb. Res. 2009, 124, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived h2o2 mediates endoplasmic reticulum signaling through local ras activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Celestino, I.; Checconi, P.; Amatore, D.; De Angelis, M.; Coluccio, P.; Dattilo, R.; Alunni Fegatelli, D.; Clemente, A.M.; Matarrese, P.; Torcia, M.G.; et al. Differential redox state contributes to sex disparities in the response to influenza virus infection in male and female mice. Front. Immunol. 2018, 9, 1747. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M.; Ostertag, D.; Li, Z.W.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. Jnk2 and ikkbeta are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef]
- Ludwig, S.; Wolff, T.; Ehrhardt, C.; Wurzer, W.J.; Reinhardt, J.; Planz, O.; Pleschka, S. Mek inhibition impairs influenza b virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561, 37–43. [Google Scholar] [CrossRef]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The rsk factors of activating the ras/mapk signaling cascade. Front. Biosci. 2008, 13, 4258–4275. [Google Scholar] [CrossRef]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza a virus ns1 protein to pkr mediates the inhibition of its activation by either pact or double-stranded rna. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef]
- Vaid, R.; Sharma, N.; Chauhan, S.; Deshta, A.; Dev, K.; Sourirajan, A. Functions of polo-like kinases: A journey from yeast to humans. Protein Pept. Lett. 2016, 23, 185–197. [Google Scholar] [CrossRef]
- Sun, D.; Luthra, P.; Li, Z.; He, B. Plk1 down-regulates parainfluenza virus 5 gene expression. PLoS Pathog. 2009, 5, e1000525. [Google Scholar] [CrossRef]
- Chen, Y.C.; Su, W.C.; Huang, J.Y.; Chao, T.C.; Jeng, K.S.; Machida, K.; Lai, M.M. Polo-like kinase 1 is involved in hepatitis c virus replication by hyperphosphorylating ns5a. J. Virol. 2010, 84, 7983–7993. [Google Scholar] [CrossRef] [PubMed]
- Bosco, R.; Melloni, E.; Celeghini, C.; Rimondi, E.; Vaccarezza, M.; Zauli, G.J.M. Fine tuning of protein kinase c (pkc) isoforms in cancer: Shortening the distance from the laboratory to the bedside. Mini Rev. Med. Chem. 2011, 11, 185–199. [Google Scholar] [CrossRef]
- Hrincius, E.R.; Dierkes, R.; Anhlan, D.; Wixler, V.; Ludwig, S.; Ehrhardt, C. Phosphatidylinositol-3-kinase (pi3k) is activated by influenza virus vrna via the pathogen pattern receptor rig-i to promote efficient type i interferon production. Cell. Microbiol. 2011, 13, 1907–1919. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic ras/mapk and survival pi3k/akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The ras-erk and pi3k-mtor pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Ali, K.; Bilancio, A.; Geering, B.; Foukas, L.C. Signalling by pi3k isoforms: Insights from gene-targeted mice. Trends Biochem. Sci. 2005, 30, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A.; Hale, B.G. Influenza a viruses and pi3k: Are there time, place and manner restrictions? Virulence 2012, 3, 411–414. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza a virus ns1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Ludwig, S. Activation of phosphatidylinositol 3-kinase signaling by the nonstructural ns1 protein is not conserved among type a and b influenza viruses. J. Virol. 2007, 81, 12097–12100. [Google Scholar] [CrossRef] [PubMed]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smallwood, H.S.; Duan, S.; Morfouace, M.; Rezinciuc, S.; Shulkin, B.L.; Shelat, A.; Zink, E.E.; Milasta, S.; Bajracharya, R.; Oluwaseum, A.J.; et al. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep. 2017, 19, 1640–1653. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.L.; Cronin, J.G.; Noleto, P.G.; Sheldon, I.M. Glucose availability and amp-activated protein kinase link energy metabolism and innate immunity in the bovine endometrium. PLoS ONE 2016, 11, e0151416. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.C.; Hardie, D.G. Amp-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Winder, W.W.; Hardie, D.G. Amp-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol.-Endocrinol. Metab. 1999, 277, E1–E10. [Google Scholar] [CrossRef]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. Ampk exerts dual regulatory effects on the pi3k pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ulk1 (atg1) phosphorylation of sting to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Prantner, D.; Perkins, D.; Vogel, S. Ampk regulates innate immune signaling and viral control through ulk-1 dependent decrease of sting expression (inm7p.430). J. Immunol. 2014, 192, 123.8. [Google Scholar]
- Prantner, D.; Perkins, D.J.; Vogel, S.N. Amp-activated kinase (ampk) promotes innate immunity and antiviral defense through modulation of stimulator of interferon genes (sting) signaling. J. Biol. Chem. 2017, 292, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Dumit, V.I.; Dengjel, J. Autophagosomal protein dynamics and influenza virus infection. Front. Immunol. 2012, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Vallerie, S.N.; Hotamisligil, G.S. The role of jnk proteins in metabolism. Sci. Transl. Med. 2010, 2, 60rv65. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Jain, K.; Basu, A. Regulation of autophagy by kinases. Cancers 2011, 3, 2630–2654. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded rna-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Lee, Y.J. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem. J. 2003, 373, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. Mapk signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef]
- Maines, M.D. Biliverdin reductase: Pkc interaction at the cross-talk of mapk and pi3k signaling pathways. Antioxid. Redox Signal. 2007, 9, 2187–2196. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in nf-κb signaling pathways. Nat. Immunol. 2011, 12, 695. [Google Scholar] [CrossRef]
- Zubair, H.; Azim, S.; Srivastava, S.K.; Ahmad, A.; Bhardwaj, A.; Khan, M.A.; Patel, G.K.; Arora, S.; Carter, J.E.; Singh, S.; et al. Glucose metabolism reprogrammed by overexpression of ikk-epsilon promotes pancreatic tumor growth. Cancer Res. 2016, 76, 7254–7264. [Google Scholar] [CrossRef]
- Tornatore, L.; Thotakura, A.K.; Bennett, J.; Moretti, M.; Franzoso, G. The nuclear factor kappa b signaling pathway: Integrating metabolism with inflammation. Trends Cell Biol. 2012, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Invest. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C. The tail wagging the dog—Regulation of lipid metabolism by protein kinase C. FEBS J. 2013, 280, 5371–5383. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of pi3k/akt/mtor signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of inhibiting pi3k-akt-mtor pathway on lipid metabolism homeostasis in goose primary hepatocytes. Animal 2016, 10, 1319–1327. [Google Scholar] [CrossRef]
- Li, C.J.; Jiang, C.; Liu, Y.; Bell, T.; Ma, W.; Ye, Y.; Huang, S.; Guo, H.; Zhang, H.; Wang, L.; et al. Pleiotropic action of novel bruton’s tyrosine kinase inhibitor bgb-3111 in mantle cell lymphoma. Mol. Cancer Ther. 2018, 18, 267–277. [Google Scholar] [CrossRef]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. Tgfbeta-induced emt requires focal adhesion kinase (fak) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef]
- Lim, S.T.; Miller, N.L.; Chen, X.L.; Tancioni, I.; Walsh, C.T.; Lawson, C.; Uryu, S.; Weis, S.M.; Cheresh, D.A.; Schlaepfer, D.D. Nuclear-localized focal adhesion kinase regulates inflammatory vcam-1 expression. J. Cell Biol. 2012, 197, 907–919. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of fak kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Calalb, M.B.; Polte, T.R.; Hanks, S.K. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: A role for src family kinases. Mol. Cell. Biol. 1995, 15, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Genna, A.; Gil-Henn, H. Fak family kinases: The yin and yang of cancer cell invasiveness. Mol. Cell. Oncol. 2018, 5, e1449584. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Delorme-Axford, E.; Harris, K.; Morosky, S.; Ikizler, M.; Dermody, T.S.; Sarkar, S.N.; Coyne, C.B. Focal adhesion kinase is a component of antiviral rig-i-like receptor signaling. Cell Host Microbe 2012, 11, 153–166. [Google Scholar] [CrossRef]
- Dwyer, S.F.; Gao, L.; Gelman, I.H. Identification of novel focal adhesion kinase substrates: Role for fak in nfkappab signaling. Int. J. Biol. Sci. 2015, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M. Targeting focal adhesion kinase in cancer-part i. Anticancer Agents Med. Chem. 2010, 10, 713. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Focal adhesion kinase as a cancer therapy target. Anticancer Agents Med. Chem. 2010, 10, 735–741. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Editorial: Focal adhesion kinase signaling in cancer--part ii. Anticancer Agents Med. Chem. 2011, 11, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of fak in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (fak) inhibitor, targeting y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits fak autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar]
- Golubovskaya, V.M.; Nyberg, C.; Zheng, M.; Kweh, F.; Magis, A.; Ostrov, D.; Cance, W.G. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 2008, 51, 7405–7416. [Google Scholar] [CrossRef]
- O’Brien, S.; Golubovskaya, V.M.; Conroy, J.; Liu, S.; Wang, D.; Liu, B.; Cance, W.G. Fak inhibition with small molecule inhibitor y15 decreases viability, clonogenicity, and cell attachment in thyroid cancer cell lines and synergizes with targeted therapeutics. Oncotarget 2014, 5, 7945–7959. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Curtin, L.; Groman, A.; Sexton, S.; Cance, W.G. In vivo toxicity, metabolism and pharmacokinetic properties of fak inhibitor 14 or y15 (1, 2, 4, 5-benzenetetramine tetrahydrochloride). Arch. Toxicol. 2015, 89, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Back to Top
Inga kommentarer:
Skicka en kommentar